
パーキンソン病・多系統萎縮症グループ
研究内容
パーキンソン病はアルツハイマー病に次いで患者数の多く、疾患修飾治療(根治治療)がまだ存在しない神経難病です。病理学的にはαシヌクレインを主な構成成分とするLewy小体の形成を特徴としますが、病態の詳細はまだ明らかとはなっておらず、病因タンパク質とされるαシヌクレインの凝集、タンパク質の輸送・分解系の障害、ミトコンドリアの機能・品質管理障害や神経炎症などが重要な病態と考えられています。また、多系統萎縮症は、多くは40歳以降に孤発性に発症し、臨床的にはパーキンソニズム、小脳失調、自律神経障害を呈する神経難病です。病理学的には、パーキンソン病と同じくαシヌクレインの凝集体を認めますが、パーキンソン病の凝集とは立体構造が異なった、多系統萎縮症に特異的な構造をしており、神経細胞ではなく、オリゴデンドロサイト主体に認めます(glial cytoplasmic incusion: GCIと呼ばれます)。病態として、GCIの形成の他、オリゴデンドロサイトの前駆体からの分化成熟障害、鉄代謝異常や著明な鉄沈着をきたすことがこれまで明らかとなっていますが、その病態はまだ多くの部分が未解明で、パーキンソン病と同様に、疾患修飾治療もありません。私たちの研究室では、これらの疾患を中心とした以下のような研究開発を行っています。
パーキンソン病の病態解明と治療法の開発
培養細胞や、マウス・ラットなどのげっ歯類、小型魚類だけでなく、よりヒトに近いモデルとして小型霊長類であるマーモセットなど様々な動物を用いて、特に前駆期を含めたパーキンソン病の全自然史を再現するようなモデル動物の開発を行っています。
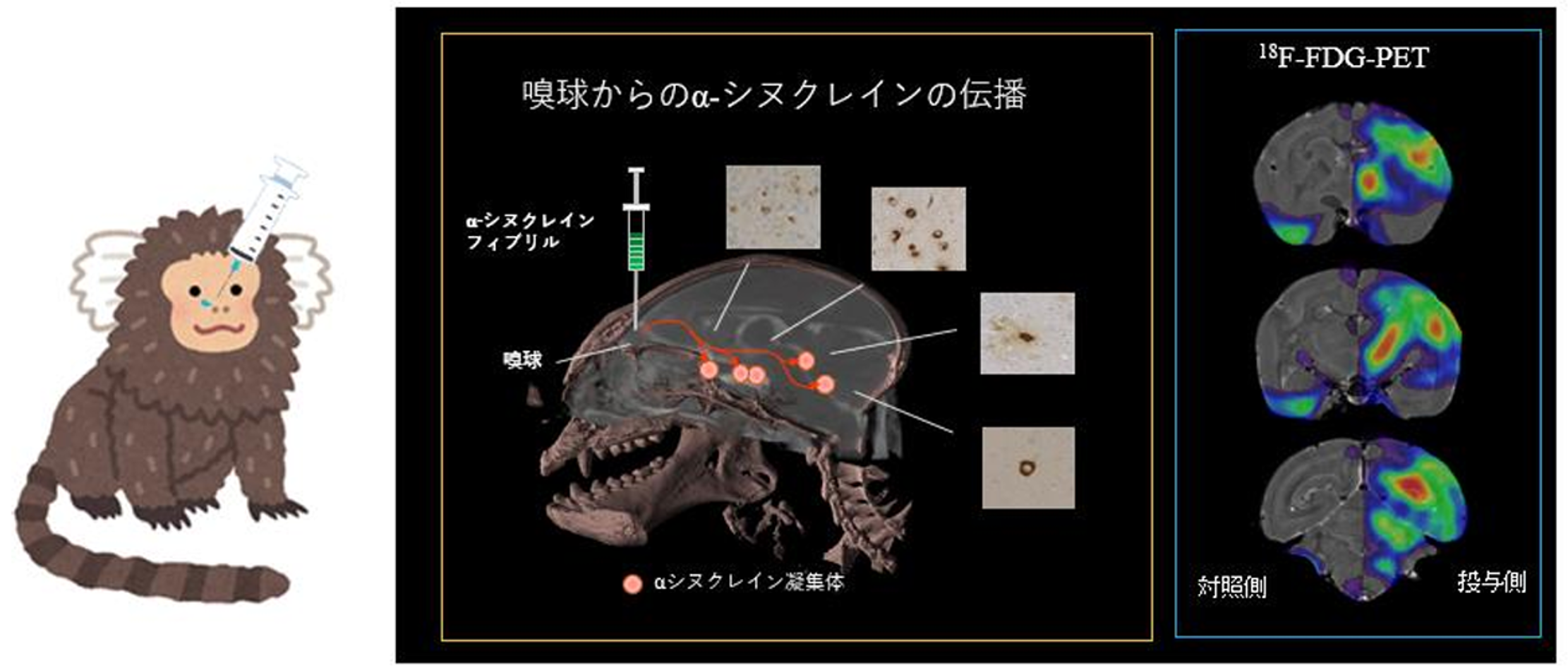
例えば、孤発性パーキンソン病のリスク遺伝子を用いた遺伝子改変モデルとしては、パーキンソン病で最も重要病因タンパク質であるαシヌクレインの遺伝子を導入したαシヌクレインBACトランスジェニックマウス・ラットや、パーキンソン病の最大のリスク因子であるGBA変異を持ちαシヌクレイン凝集と経時的な神経細胞死を呈するメダカモデルを作製しました(Uemura et al. PLoS genet. 2015)。特に、当研究室のαシヌクレインBACトランスジェニックマウスは、レム睡眠行動異常症(RBD)や嗅覚低下などのヒトと類似のパーキンソン病の前駆期症状を呈するモデルとして、世界中で高く評価され注目されています(Taguchi et al. Brain 2019)。
また、パーキンソン病の病態進展モデルとして注目されているBraak仮説に基づき、最初期病変からのαシヌクレインの伝播を再現するモデルの作製を試みています。これまでに、αシヌクレイン線維の嗅球・鼻腔あるいは消化管へ投与し伝播するマウス・マーモセットモデルを作製し報告してきました(Uemura et al. Mol Neurodegener. 2018, Uemura et al. Mov Disord. 2021)。例えば、αシヌクレインの凝集体をマーモセットの嗅球へ投与すると、凝集体が脳内を伝播し、さらに18F-FDG-PETを用いて調べると、パーキンソン病の進行期でみられるような広範な脳機能低下が生じることが分かりました(Sawamura et al. Mov Disord. 2022)。
さらにこれらのモデルを用いて神経系のみならず全臓器に注目して解析することで、病態解明のみならず、バイオマーカー開発や治療候補薬物の検証を行い実臨床への還元を目指しています。
これまでに、抗てんかん薬として用いられているペランパネルがαシヌクレインの伝播を抑制する可能性があることを見出しました(Ueda et al. Move Disord 2021)。
多系統萎縮症の病態解明と治療法の開発
オリゴデンドロサイトへの疾患特異的なαシヌクレインの凝集、オリゴデンドロサイトの前駆体からの分化成熟障害、鉄代謝異常や著明な鉄沈着を生じるといった知見に基づいたモデル動物の作製を行っています。例えばこれまでに、オリゴデンドロサイト特異的にαシヌクレインを発現させることで、オリゴデンドロサイトへのαシヌクレインの凝集をモニターできるマウスを作製しました(Ishimoto et al. Mol Brain. 2024)。また、パーキンソン病と多系統萎縮症の疾患特異的αシヌクレイン線維に着目し、パーキンソン病と多系統萎縮症のそれぞれに疾患特異的なαシヌクレイン線維を用いたマウス・マーモセットモデルの開発と、各線維が惹起する病態解明の研究を行っています(Uemura et al Nat Commun. 2023)。
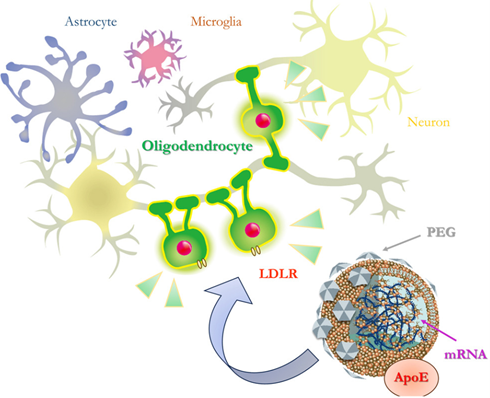
さらには治療開発として、米国アクチュラス社と共同研究で、オリゴデンドロサイト特異的なリポソームを見出し、オリゴデンドロサイトにmRNAを高効率で導入できることを報告しました(Sawamura et al. Mol Ther Nucleic Acids. 2024)。このリポソームはApoEと結合し、LDLRを介してオリゴデンドロサイト特異的に取り込まれます。そして、これまでに、オリゴデンドロサイトが障害される先天性疾患であるクラッベ病のモデルマウスの症状を改善できることを示しました。この成果は多系統萎縮症の治療にも結び付くものとして研究を進めています。
バイオマーカー開発
神経変性疾患では、診断時には既に一定以上の神経変性をきたしていることが知られています。例えば、パーキンソン病では運動症状が現れた時点で、黒質のドパミン細胞が半分以下になっていることがわかっています。このため、これらの疾患の疾患修飾薬の開発には超早期あるいは前駆期での診断が重要です。また、臨床試験においては、凝集する異常タンパク質をターゲットにして開発されている治療薬が多く、正確な凝集タンパク質の評価が必要といえます。さらには、変性は年余をかけて進行することから、治療開発にかかるコストは時間的にも膨大になるため、疾患進行予測や治療効果判定ができるバイオマーカーの開発も急務といえます。しかしながら、いずれにも利用できるバイオマーカーは、まだ実用化にはいたっていません。私たちの研究室では、多くの患者さんや健康な方に御協力をいただき、ELISA、RT-QUIC、特殊なbiosensorなどの様々なmodalityを用いた体液サンプル、画像データ、組織サンプルからこれらを可能とするバイオマーカーの開発を行っています。例えば、プロテオミクス解析と機械学習を用いて、脳脊髄液のプロテオソームの変化が疾患の診断・予後予測に有用であることを見出しました(Tsukita et al. Neurology 2024)。
Integrated Bio-Clinical Informatics and Translational Research(→独自Webページはこちら)
Integrated Bio-Clinical Informatics and Translational Research Teamでは、オープンデータ、臨床コホート、研究室で独自に生成したオミクスデータを統合し、神経疾患の臨床的・生物学的理解を深めることを目指しています。クリニカルビッグデータ、画像、体液オミクス、single-cell解析などに先進的なインフォマティクス手法を応用し、診断・予後予測・治療標的探索につながる知見の創出に取り組んでいます。
これまでに、脳脊髄液プロテオミクスと機械学習を用いてパーキンソン病の発症・進行に関わるタンパク質シグネチャーを同定したほか、APOE ε4を有するパーキンソン病患者において血中リンパ球数が認知機能低下を予測しうること、日常の身体活動量や運動習慣が長期的な症状進行に抑制的に働くことを報告してきました。また、運動予備能、患者報告アウトカム、脳梗塞後オリゴデンドロサイト前駆細胞の低酸素応答、移植医療におけるHLA分子ミスマッチ解析など、臨床と生物学を横断するデータ駆動型研究を展開しています。
パーキンソン病・多系統萎縮症グループ メンバー
脳神経内科
中西悦郎(助教)
田口智之(特定助教)
石本智之(特定病院助教)
月田和人(特定講師 → Integrated Bio-Clinical Informatics and Translational Research Team )
平藤哲也(特定助教)
孝橋睦生(D4)
成宮悠爾(D4)
櫻井靖久(D3)
上田明広(D2)
常嘉瑞(研究生)
大平純一朗(客員研究員)
多系統萎縮症治療学講座
松澤秀一(特定准教授)
山門穂高(特定准教授)
向敦史(プロジェクト研究員)
酒巻和弘(非常勤研究員)
大野美樹(非常勤研究員)
上村紀仁(非常勤研究員・大阪公立大学)
その他
髙橋良輔(特定教授・総合研究推進本部)
生野真嗣(講師・医学教育・国際推進センター)